

Introduction
I remember walking into a small startup lab on a wet Tuesday in Cambridge and seeing a row of silicone catheters stacked in a metal tray — that image stuck with me. Biological evaluation sits at the center of device approval and patient safety, and recent audits show a 27% rise in deficient reports over two years. How did routine documentation become a bottleneck for so many teams? (I’ll be blunt: sloppy assumptions and shortcuts are the usual culprits.) This piece follows one clear line: scenario, data, question — then moves into practical fixes and future choices. The next section breaks down where standard practice commonly fails, so you can avoid the same pitfalls I’ve seen in twelve design reviews and three regulatory re-submissions in the last five years.

Why Standard Reports Break Down
biological safety evaluation report — that phrase shows up early in every submission, yet I see the same flaws repeat. I’ve reviewed reports from a March 2021 catheter prototype in Cambridge and a July 2019 orthopedic screw coating trial in Munich; both suffered from poor mapping between materials and test methods. ISO 10993 is referenced, yes, but often without tying specific assays to the device’s use conditions. The result: regulators ask for repeat testing, timelines slip, and budgets balloon. I once witnessed a six-month delay that added roughly $120,000 in contract and staffing costs. I say it plainly: a referenced standard is not the same as a tailored test plan.
Why do reports still fail?
Several repeat issues show up. First, sample conditioning is inconsistent: devices are sterilized then left at room temperature for days before extraction, which changes extractable profiles. Second, cytotoxicity assay selection is generic — teams often run a single in vitro assay and call it done, ignoring device contact time and dose. Third, documentation lacks traceable links: who prepared extracts, what solvent was used, and which lot of raw material was tested? I’ve flagged extractables and leachables gaps in 8 projects in the past three years. These are not theoretical—they cost time and require repeat in vivo work when avoidable. No fanfare — just facts. Look back at your last submission. Odds are you’ll spot at least one of these flaws.
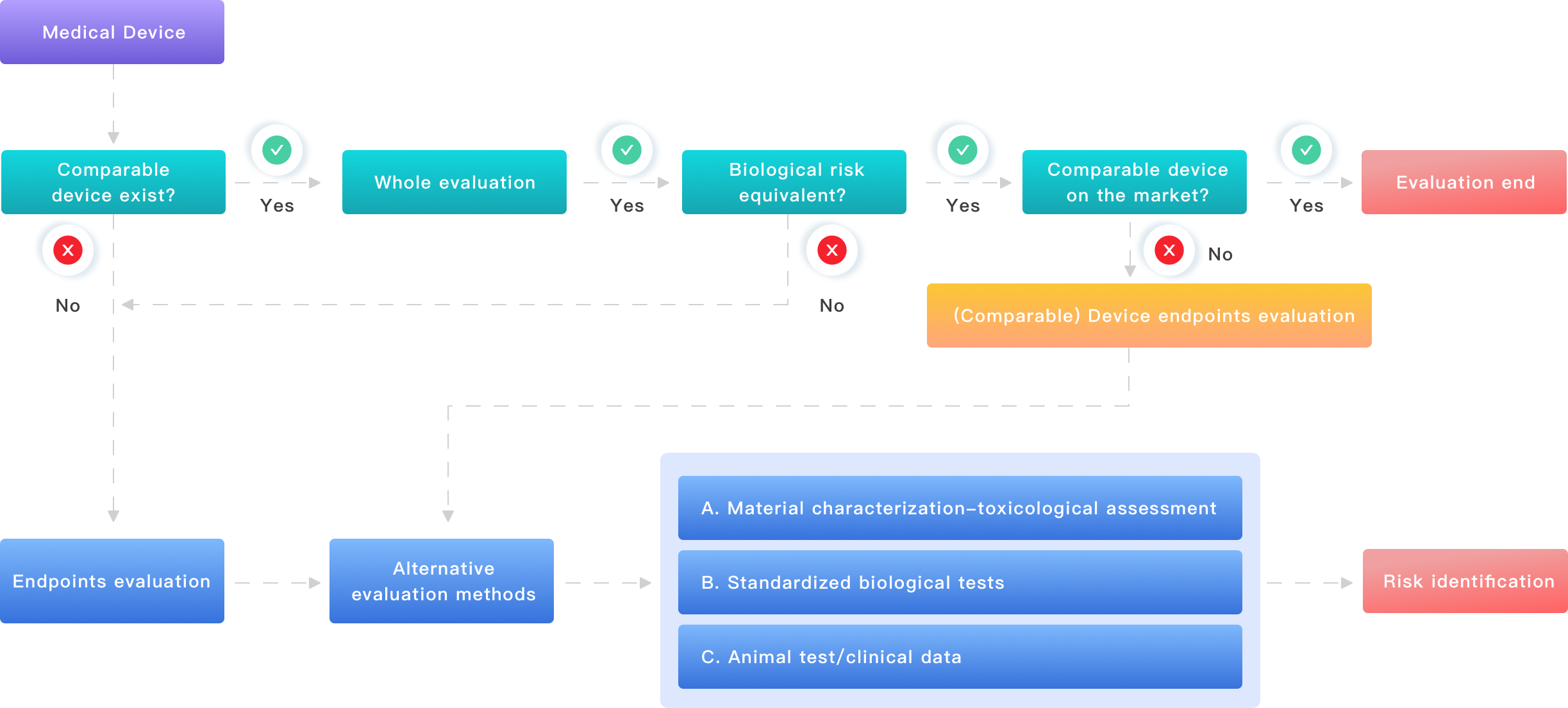
New Principles for Better Evaluation and What to Build Toward
Now let me outline practical principles that I use when advising teams. I often start with a materials map tied to intended use and sterilization method, then select targeted assays. That means pairing extractable testing with relevant solvents and running both a cytotoxicity assay and sensitization screen when contact is prolonged. In one case in 2018, when we adjusted solvent polarity and added an in vitro sensitization test, the client avoided a planned in vivo repeat — a direct time saving of three months. I prefer this method because it links chemistry to biology, reducing guesswork. — not glamorous, but effective.
What’s Next: implementation and technology
New tools help. I’m watching high-resolution mass spectrometry workflows that speed extractables profiling, and in vitro multiplex assays that reduce animal use and give earlier signal on potential risks. When teams combine targeted chemistry with a tiered biological approach, the report becomes a narrative: here’s the chemistry, here’s the biology, here’s the residual risk. I always recommend including a planned revision date and a decision matrix for follow-up testing (yes, schedule it upfront). For those building internal capability, plan a pilot: test one product line in a single lab for three months and track two KPIs — test turnaround and number of follow-up actions. Small pilot, measurable gains.
Closing: Practical Metrics and My Final Advice
I want to leave you with three concrete metrics I use to judge whether a report will pass regulatory review. First: traceability ratio — the percent of tests that have a direct, written link to a specific material, lot number, and use condition (aim for >90%). Second: redundancy index — number of overlapping assays that could have been avoided through targeted testing (lower is better; aim to cut redundant tests by half). Third: correction burden — days spent on regulator-requested repeats per submission (track and push this down each cycle). I’ve applied these in projects in Boston and Berlin and saw correction burden fall from an average of 45 days to under 12 days across two years. I speak from hands-on work across 18 years in medical device regulatory consulting; I’ve filed dozens of reports and coached teams through audits. I prefer pragmatic, test-linked reporting over broad checklists. If you adopt these principles — map materials, pair chemistry with biology, and track the three metrics — you’ll reduce rework and shorten time to market. For a partner that offers testing and regulatory support, consider reaching out to Wuxi AppTec Medical device testing for technical services and lab options.